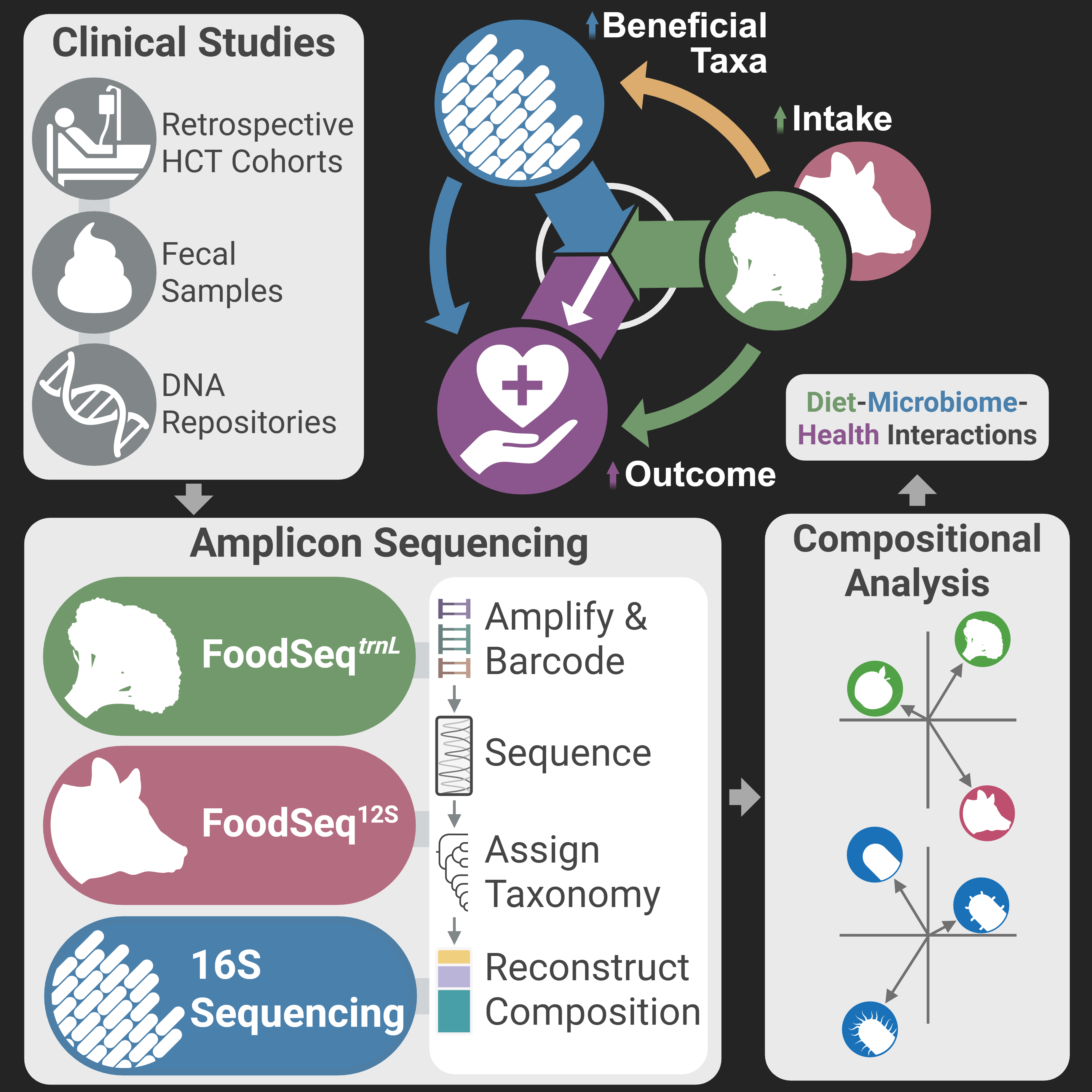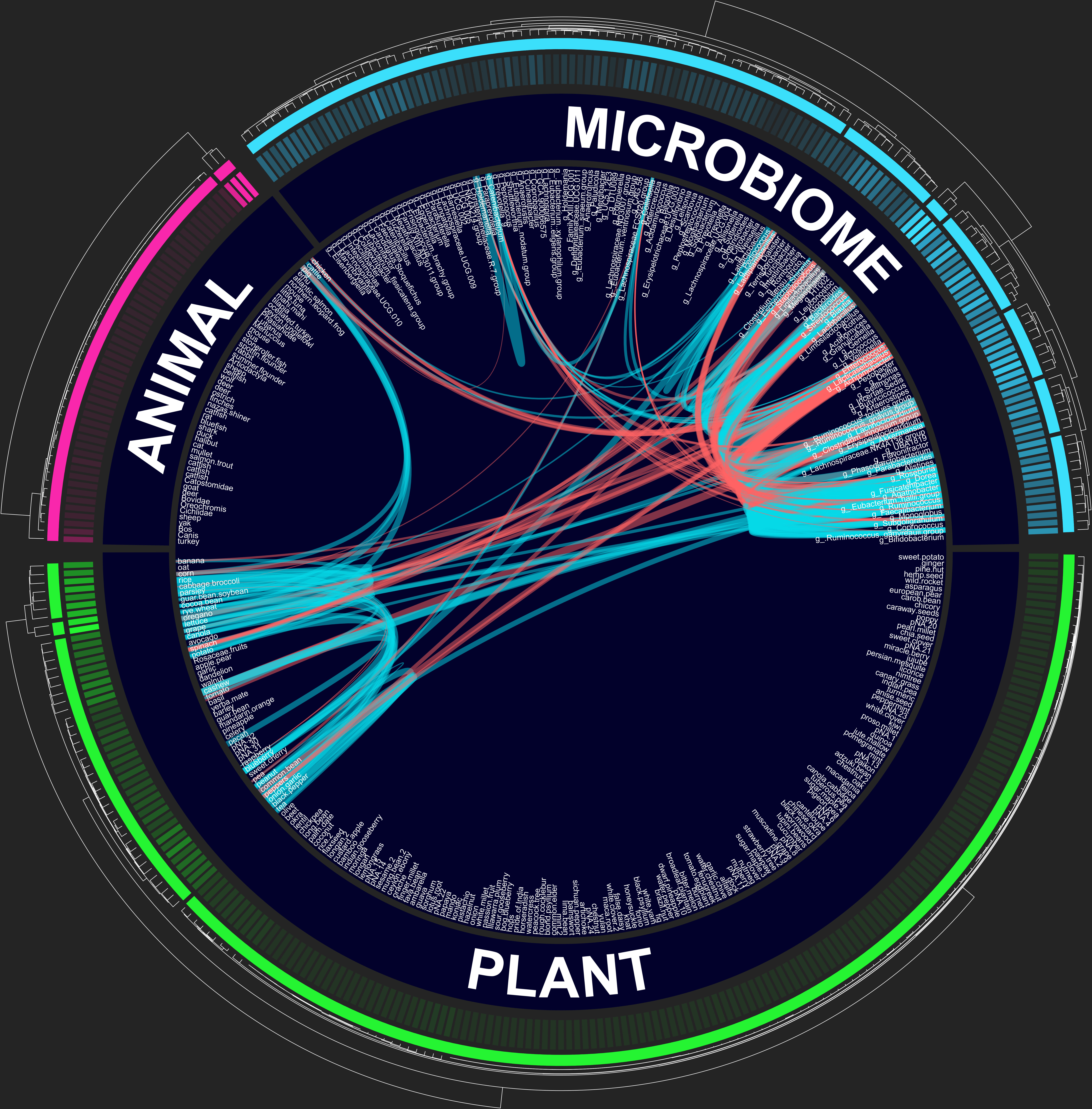




Molecular Biology & Genetics
Next-generation sequencing (NGS)
Bacterial 16S
Plant trnL
Animal 12S
Sequencing library construction
Molecular cloning
Overlap extension PCR
Restriction digest
λ-Red recombination
Gibson assembly
Di-/tri-parental conjugation
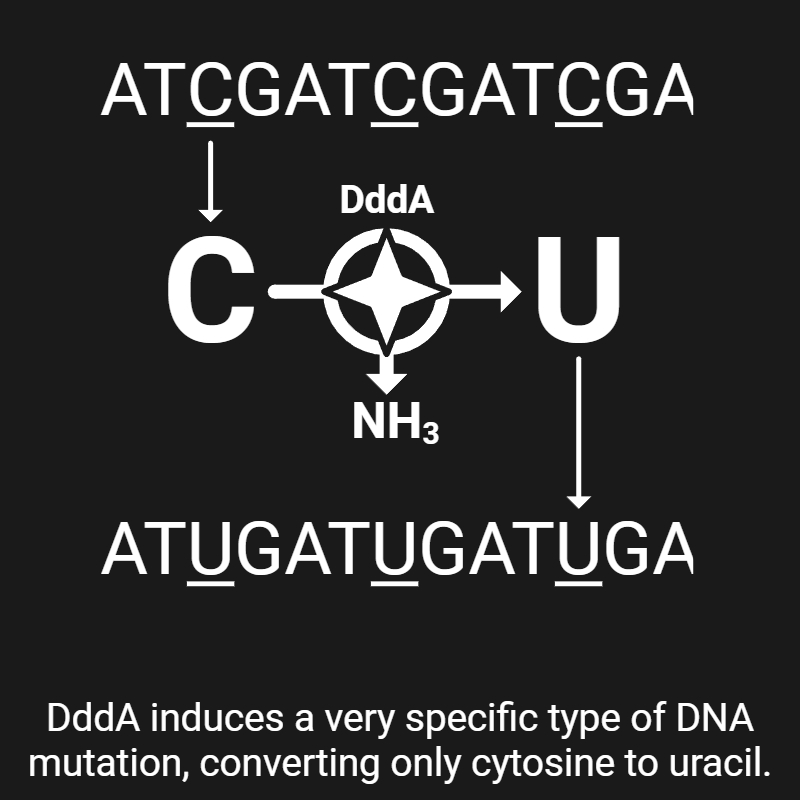
Bacterial mutagenesis
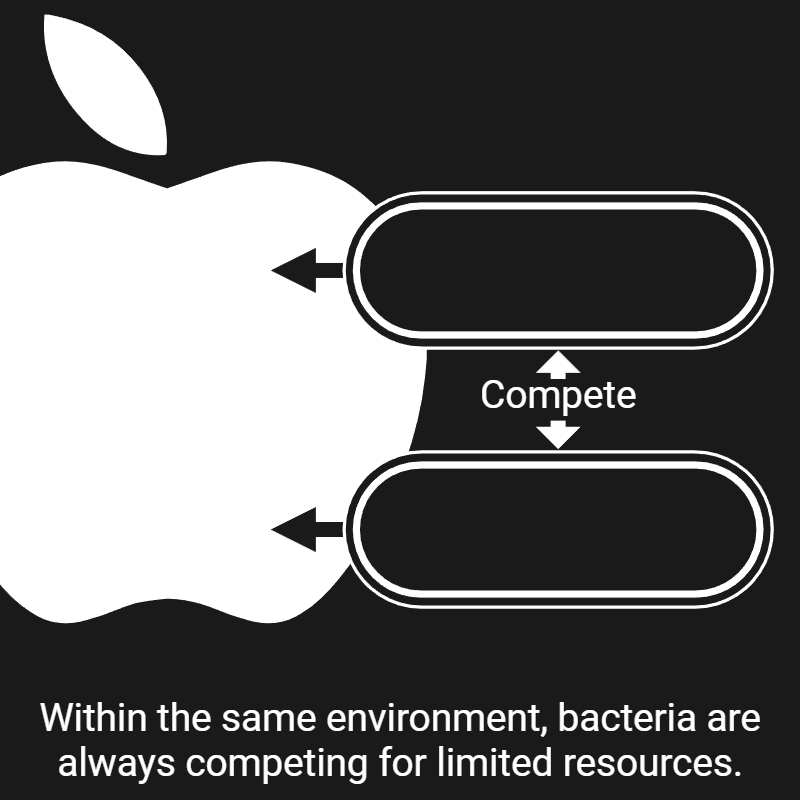
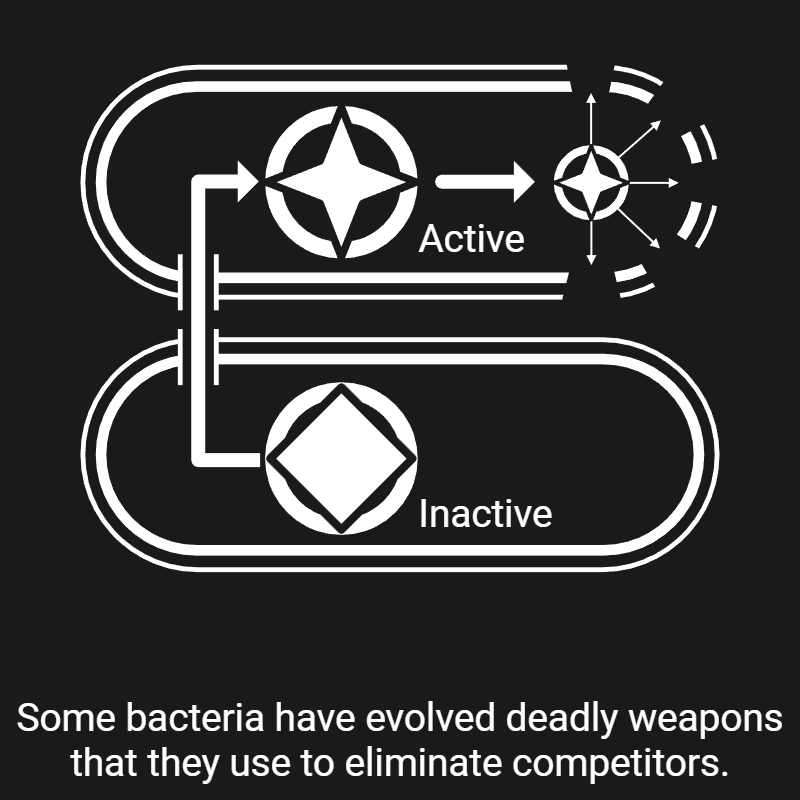
Bacterial competition
DNA extraction
PCR/qPCR
Plasmid design
Primer design
Protein Biochemistry
Immobilized metal affinity chromatography
Protein expression
SDS-PAGE
ELISA
Automation & Instruments
MiniSeq (Illumina)
Anaerobic chamber (Coy)
GC-FID (Agilent)
HPAE (Agilent)
Lyophilization (Labconco)
Automatic liquid handling (epMotion)